
Räumlich-zeitlich aufgelöste Einzelzell-Bioinformatik
Dr. Fabian Kern
Infektionen und Therapien mit Antiinfektiva bewirken verschiedenste, räumlich und zeitlich abhängige biochemische Prozesse mit vielen noch zu entdeckenden und oft zelltypabhängigen Mechanismen. Wir entwickeln fortschrittliche bioinformatische Ansätze, wie z. B. domänenspezifische neuronale Netze auf der Grundlage von räumlich-zeitlichen RNA-Profilen, für die rasche und verständliche Analyse gesunder und kranker Zellfunktionen. Neue Computermodelle ermöglichen es uns, ein tieferes mechanistisches Verständnis zellulärer Signalkaskaden und -wege bei Infektionskrankheiten zu erlangen. Auf diese Weise unterstützen wir von der bioinformatischen Seite den Translationsprozess für die am HIPS entwickelten Wirkstoffe, in Richtung erfolgreicher klinischer Anwendungen und neuen Therapien.
Unsere Forschung
Hochdurchsatz-Sequenzierung und hocheffiziente Bioinformatiksoftware sind der Motor der meisten Forschungs- und Industriezweige in der modernen Biomedizin. Wesentliche Faktoren der bakteriellen oder viralen Pathogenität, z.B. Virulenz oder Mechanismen der Arzneimittelresistenz, werden von komplexen Genregulationswegen sowie von der Art der Wirtszelle und des Erregers bestimmt und können mithilfe von Sequenzierung beleuchtet werden. Die zugrundeliegenden molekularen Prozesse, die zu den beobachteten klinischen Phänotypen führen, können dann mithilfe von KI- und bioinformatischen Methoden entschlüsselt werden. In der laufenden Forschung zu Antibiotika, Antiinfektiva und Resistenzmechanismen werden zunehmend räumlich und zeitlich aufgelöste Einzelzelldaten verwendet, die uns vielversprechende neue intra- oder extrazelluläre „Targets“ von Interesse offenbaren.
Die von Dr. Fabian Kern geleitete Forschungsgruppe hat sich zum Ziel gesetzt, solche Targets mit Hilfe von Deep Learning und Big-Data-Analytics-Techniken zu identifizieren. Dabei werden unter anderem von Labor-Infektionsmodellen und klinischen Proben gewonnene Daten verwendet, die in enger Zusammenarbeit mit Partnern aus dem HIPS, der Medizinischen Fakultät sowie der Naturwissenschaftlichen Fakultät der Universität des Saarlandes erhoben werden. Im Einzelnen geht es uns darum, die Infektionsreaktionen des Wirtes an wichtigen biologischen Schnittstellen wie dem Darm oder der Blut-Hirn-Schranke besser zu verstehen. Unser langfristiges Ziel ist es, mit Hilfe von interpretierbaren Modellen des Maschinellen Lernens und graph-basierten Algorithmen diejenigen zellulären Veränderungen über Raum und Zeit zu identifizieren, die ursprünglich durch Infektionen verursacht werden. Zu diesem Zweck kontextualisieren wir molekulare Datensätze mit demografischen, klinischen und individuellen Merkmalen sowie Informationen über Wirkstoffe. Auf diese Weise versuchen wir schließlich, ein besseres Verständnis komplexer menschlicher Phänotypen wie vorzeitiges Altern oder chronische Krankheiten älterer Menschen zu ermöglichen, die zum Teil mit einzelnen, vorangegangenen Phasen schwerer Infektionen zusammenhängen können.
Team-Mitglieder

Dr. Fabian Kern
Gruppenleiter

Matthias Flotho
Doktorand
Forschungsprojekte
Maschinelles Lernen anhand modernster räumlicher und Einzelzell-Transkriptomik
Die rasante Entwicklung von (RNA-)Sequenzierungstechnologien mit beispiellosem Durchsatz und hoher Auflösung ermöglicht es uns, detaillierte molekulare Profile für Millionen einzelner Zellen aus menschlichem Gewebe oder Körperflüssigkeiten und sogar für einzelne krankheitsverursachende Keime zu erfassen. Neuste experimentelle Ansätze, bei denen Methoden zur individuellen räumlichen Markierung von RNA-Molekülen aus einzelnen Zellen für die anschließende Hochdurchsatz-Sequenzierung kombiniert werden, eröffnen eine Vielzahl potenzieller Möglichkeiten der Untersuchung zellulär-räumlicher Muster bei Infektionskrankheiten (Spatial Transcriptomics). Für eine tiefere Analyse der Sequenzierungsdaten und die Entdeckung funktioneller Zusammenhänge sind fortgeschrittene integrative Werkzeuge aus der Bioinformatik erforderlich. Darüber hinaus spielen einige bekannte, aber auch bisher verborgene technische Faktoren eine wichtige Rolle bei der Datengenerierung und beeinflussen daher immer unser Vorgehen zur maschinellen Interpretation der Ergebnisse. Deshalb nutzen wir in enger Zusammenarbeit mit führenden Unternehmen auf diesem Gebiet verschiedene hochmoderne Technologielösungen (z.B. Visium, STOmics), um unsere Modelle und Programme unter ganz verschiedenen Versuchsbedingungen zu testen. In erster Linie sind wir auf der Suche nach interessanten und anspruchsvollen Anwendungen in den Bereichen Infektion, Neurodegeneration und Antibiotika-Forschung, um letztlich die Kartographie zellulärer und molekularer Merkmale als Grundlage für neue Zielmoleküle zu verbessern.

Entwicklung graph-basierter Methoden zur Vorhersage zellulärer Nachbarschaften und Signalwege
Um die Generierung von KI-fähigen und mit maschinellem Lernen kompatiblen Datensätzen zu erleichtern, nutzen wir die am und um das HIPS herum verfügbare Expertise sowie das vorhandene Fachwissen, um unseren experimentellen Daten in Modelle zu abstrahieren. Zu diesem Zweck suchen wir nach neuen computergestützten Ansätzen, z. B. Hashing, um in großen Datensätzen schnell zwischen pathogenen und gesunden Zellprofilen unterscheiden und einzigartige Krankheitssignaturen erkennen zu können. Die Modellierung von Zellen oder Zellgemeinschaften als Knoten in größeren Graphen ist ein vielversprechender Ansatz, der in letzter Zeit Aufmerksamkeit auf diesem Gebiet erhalten hat. Die Kanten zwischen den Zellknoten werden zum Beispiel durch Liganden-Rezeptor-Wege oder andere biochemische Prozesse bestimmt, die typischerweise durch die Genexpression gesteuert werden. Unsere Methoden zielen darauf ab, reproduzierbare und testbare Gensignaturen (so genannte Panels) zu entdecken, die diejenigen Teile des Transkriptoms, d.h. der zellulär verfügbaren RNA zu entschlüsseln, die spezifisch von Krankheitserregern beeinflusst werden. Im Prinzip können solche Panels dann auch verwendet werden, um den Wirkmechanismus eines bestehenden oder neuen Wirkstoffs zu beurteilen, welcher im Idealfall zumindest bis zu einem gewissen Grad degenerierte Genregulationsprogramme in einen gesunden und homöostatischen Zustand umkehrt.
Identifizierung verwertbarer Krankheitsmarker an biologischen Schnittstellen zur Gewinnung neuer pharmazeutischer Wirkstoffe
Biologische Barrieren sind ein wesentliches Mittel des menschlichen Körpers zur Herstellung bestimmter physiologischer Bedingungen und spielen eine wichtige Rolle für unser Immunsystem. In erster Linie halten sie einzelne Gewebekompartimente voneinander getrennt und verschiedene Bereiche steril, d.h. frei von zirkulierenden Mikroben. Es gibt zahlreiche Beispiele für menschliche Krankheiten, bei denen gestörte biologische Schnittstellen bzw. Barrieren eine der Hauptursachen für den Eintritt von Krankheitserregern sind. Gleichzeitig besteht die ständige Herausforderung bei der Entwicklung von Arzneimitteln darin, kleine oder eben natürlich abgeleitete, meist komplexere Wirkstoffe zu einem bestimmten Zeitpunkt und in einer bestimmten Konzentration über diese Barrieren hinweg zu transportieren. Daher sind wir daran interessiert, die Genexpressionsprogramme der oft sehr spezialisierten Zellen wie Epi- oder Endothelzellen sowie der peripheren Immunzellen, die sich an den Schnittstellen des Gewebes befinden, aufzudecken und herauszufinden, wie wir diese für die gezielte Anwendungen von Wirkstoffen nutzen können. Deshalb erkennen wir ein großes Potenzial für eine Klasse von Medikamenten, die in der Lage sind, fehlregulierte Zellen der Barrieren auf molekularer Ebene zu reparieren und so möglicherweise eine Therapie am Menschen zu unterstützen.

Implementierung von öffentlichen und frei nutzbaren wissenschaftlichen Datenbanken und Webservern
In der Bioinformatik werden routinemäßig enorme Datenmengen verschiedenster Art verarbeitet. Insgesamt gibt es einen starken Trend hin zu exponentiell steigenden Datenmengen und einer immer komplexeren Landschaft von experimentellen Techniken. Daher besteht in unserer wissenschaftlichen Gemeinschaft ein dringender Bedarf an offenen, flexiblen und hocheffizienten Formaten für die Speicherung sowie den Austausch von biomedizinischen Daten. Unsere Gruppe folgt dem FAIR-Prinzip und stützt sich auf unsere bisherigen Erfahrungen bei der Entwicklung von weit verbreiteten und von Experten begutachteten Online-Ressourcen wie etwa Datenbanken und Webservern. Wir stellen der wissenschaftlichen Gemeinschaft kontinuierlich neue Software und experimentelle Datensätze zur Verfügung und legen Wert darauf, hinreichend vorverarbeitete und umfassende Datensammlungen bereitzustellen, die letztlich auch KI-kompatibel sind. Wir sprechen uns dafür aus, Transparenz und Reproduzierbarkeit in diesen Bereichen zu ermöglichen bzw. zu stärken, und investieren daher auch in die Erforschung der Frage, wie wir Software und KI in diesen, heutzutage wesentlichen Aspekten maßgeblich verbessern können. Um dieses Ziel zu erreichen, arbeiten wir ebenfalls mit unseren Helmholtz-Kollegen vom CISPA Helmholtz-Zentrum für Informationssicherheit zusammen.
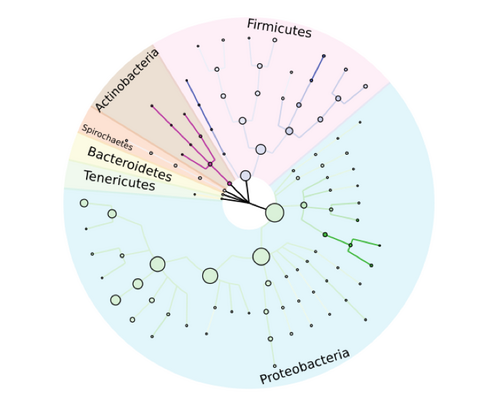
Anhand großer Plasmid-Datenbanken können detaillierte genetische Stammbäume von Bakterienstämmen erstellt werden. Abbildung adaptiert aus unserer Veröffentlichung: Georges P Schmartz, Anna Hartung, Pascal Hirsch, Fabian Kern, Tobias Fehlmann, Rolf Müller, Andreas Keller, PLSDB: advancing a comprehensive database of bacterial plasmids, Nucleic Acids Research, Volume 50, Issue D1, 7 January 2022, Pages D273–D278, https://doi.org/10.1093/nar/gkab1111. © Oxford University Press